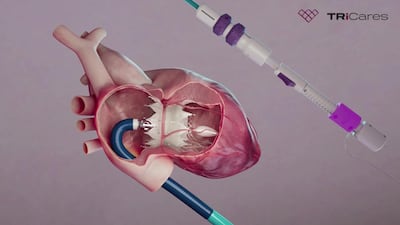
Janet Woodcock is the “right person” for the role of US Food and Drug Administration commissioner, the agency’s outgoing No. 2 official told Medtech Insight on 1 April.
“I feel very strongly about Janet Woodcock being the right person. I felt comfortable leaving my role because of the fact that Janet Woodcock is there,” said Amy Abernethy, who...